Buyer is solely responsible for determining whether Buyer has all intellectual property rights that are necessary for Buyer's intended uses of the BioLegend TotalSeq™ products. For example, for any technology platform Buyer uses with TotalSeq™, it is Buyer's sole responsibility to determine whether it has all necessary third party intellectual property rights to use that platform and TotalSeq™ with that platform.
The following protocol combines highly multiplexed protein marker detection with unbiased transcriptome profiling for thousands of single cells. Epitope detection and sample multiplexing (Cell Hashing) are enabled by BioLegend’s TotalSeq™ antibody-oligo conjugates. TotalSeq™ reagents are compatible with Cellular Indexing of Transcriptomes and Epitopes by sequencing (CITE-seq), RNA Expression And Protein sequencing assay (REAP-seq), and similar workflows.
Reagent and Instrument List
All items listed, or equivalent, are required.
- TotalSeq™ antibody-oligo conjugates
- Biotinylated antibody and oligo barcoded streptavidin
- Human TruStain FcX™ (Fc Receptor Blocking Solution) (Cat# 422301/422302)
- TruStain fcX™ (anti-mouse CD16/32) Antibody (Cat# 101319/101320)
- TruStain FcX™ PLUS (anti-CD16/32, recommended for mouse cells. BioLegend Cat# 156603)
- 8-strip PCR tubes, emulsion safe (TempAssure PCR 8-strips, USA Scientific, Cat# 1402-4700)
- Nuclease-Free Pipette Tips (e.g. ThermoFischer Scientific AM12650, AM12660 or equivalent)
- Bioanalyzer chips and reagents (DNA High Sensitivity and small RNA kit, Agilent Cat# 5067-4626 and 5067-1548)
- Quantabio sparQ PureMag Beads (Quantabio, Cat# 951960) or SPRIselect reagent (Beckman Coulter, Cat# B23317)
- E-Gel™ EX Agarose Gels, 4% (ThermoFischer Scientific Cat# G401004)
- DNA LoBind Tubes (Eppendorf, Cat# 022431021)
- PCR Thermocycler (Bio-Rad, T100™ Thermal Cycler)
|
- KAPA HiFi HotStart ReadyMix (2X) (Kapa Biosystems, Cat# KK2601) or Quantabio sparQ HiFi PCR Master Mix (2X) (Quantabio, Cat# 95192-250)
- Specific primers, see “Notes” at the end of the protocol (primers not available from BioLegend, order from preferred supplier). Hashtag primers are different than regular TotalSeq™ primers.
- Magnetic tube rack (e.g. ThermoFischer)
- Qubit (ThermoFischer Scientific, Cat# Q33226)
- Fuchs-Rosenthal Counting Chamber (Hemocytometer, VWR, Cat# 15170-230)
- Phosphate Buffered Saline (PBS) (BioLegend, Cat# 926201 or equivalent)
- Cell Staining Buffer (BioLegend, Cat# 420201)
- 80% Ethanol
- Flowmi™ Cell Strainer (Bel-Art, H-B Instrument, Cat# H13680-0040)
|
Researchers are advised to validate equivalent products when substituting the above recommendations.
Protocol
I) Cell staining for Drop-seq or 10x Genomics platforms
- Obtain all single cell suspensions from different samples/conditions that will be multiplexed in the run.
- Keep samples in separate tubes until after cell hashing and shortly before loading cells into the single cell RNA-seq instrument.
- When aiming to super-load the same sample into one run, divide the sample up into equal proportions before staining with distinct cell hashing antibodies.
- Keep cell suspensions on ice (unless otherwise stated) at all times.
- Carefully count all cells to ensure accurate quantitation.
- Make note of cell viability (>95%) and also include dead cells in the total cell count.
- If high cell death is observed, live cell enrichment (e.g. by Flow Cytometry) is recommended.
- Resuspend 1-2 million cells in 100 µl Cell Staining Buffer.
- Add 5 µl of Human TruStain FcX™ Fc Blocking reagent, or 1.0 µg of TruStain FcX™ (anti-mouse CD16/32) Antibody.
- Incubate for 10 minutes at 4°C.
- While cells are incubating in Fc Block, prepare antibody-pool using 1 µg (or titrated amounts) of each TotalSeq™ and/or hashtag or biotinylated antibody.
- To maximize performance, centrifuge the antibody pool at 14,000xg at 2 – 8°C for 10 minutes before adding to the cells.
- Carefully pipette out the liquid, avoiding the bottom of the tube, and add the TotalSeq™ antibody cocktail to the cell suspension.
- Incubate for 30 minutes at 4°C.
- Wash cells 3 times with 1 mL Cell Staining Buffer, spin 5 minutes 350g at 4°C.
*Please note that it has been observed in some cases that various factors, including cell/sample type, tube manufacturer, rotor type, wash buffer, etc., may result in an excessive number of cells coating the side of the tube. Please ensure that staining and washing conditions are appropriate for your sample type.
- If using biotinylated antibodies, incubate with the appropriate oligo barcoded streptavidin at the recommended amount specified in the product technical datasheet for 20 minutes.
- Wash cells 3 times with 1 mL of Cell Staining Buffer, spin 5 minutes 350g at 4°C.
- Resuspend cells in PBS at appropriate concentration for downstream application. (e.g. for 10x Genomics 500 cells/µl; for Drop-seq 200 cells/µl; for super-loading 1,500 cells/µl or higher).
- Filter cells through 40 µm strainers.
- Verify cell concentration by counting on hemocytometer after filtration.
- Pool all different samples/conditions at desired proportions and immediately proceed to next step.
II) Run Drop-seq (Macosko et al., 2015) or 10x Genomics single cell 3’ v2 assay as described until before cDNA amplification.
At cDNA amplification step:
Add “additive” primers to cDNA PCR to increase yield of Antibody Derived Tag (ADT, cDNA derived from the TotalSeq™antibodies) and/or Hashtag oligonucleotide (HTO, cDNA derived from the TotalSeq™ Hashtag antibodies) products:
ADT PCR additive primer (0.2 µM): 1 µl (for 10x Genomics) or 0.4 µl (for Drop-seq)
HTO PCR additive primer (0.2 µM): 1 µl (for 10x Genomics) or 0.4 µl (for Drop-seq)
Please see example table below of 10X cDNA amplification reaction table (section 2.2, Chromium Single Cell 3’ v2 Reagent Kit, Quick Reference Card (v2 Chemistry))
|
cDNA Amplification Mix
|
1X (uL)
|
|
Nuclease-Free Water
|
6
|
|
Amplification Master Mix
|
50
|
|
cDNA Additive
|
5
|
|
cDNA Primer Mix
|
2
|
|
ADT Additive Primer (0.2 µM stock)
|
1
|
|
HTO Additive Primer (0.2 µM stock)
|
1
|
|
Total
|
65
|
See notes at the end of the protocol for further details. Subtract the total volume of additive primer from the water added to the PCR reaction.
III) ADT and mRNA library preparation
A) After cDNA amplification: Separate ADT and HTO-derived cDNAs (180bp) and mRNA-derived cDNAs (>300bp).
1. Perform sparQ or SPRI selection to separate mRNA-derived and antibody-oligo-derived cDNAs.
2. Do not discard supernatant from 0.6X sparQ or SPRI. This contains the ADTs and hashtags.
- Add 0.6X sparQ or SPRI (60 µl, based on 100 µl sample volume) to cDNA reaction as described in 10x Genomics or Drop-seq protocol.
- Incubate 5 minutes and place on magnet.
- Supernatant contains ADTs and hashtags.
- Beads contain full length mRNA-derived cDNAs.
B) mRNA-derived cDNA >300bp (beads fraction).
Proceed with standard 10x Genomics or Drop-seq protocol for cDNA sequencing library preparation.
C) ADTs and/or hashtags 180bp (supernatant fraction).
1) Purify ADTs using two 2X sparQ or SPRI purifications per manufacturer protocol
- Add 1.4X sparQ or SPRI (140 µl, based on 100 µl sample volume) to supernatant to obtain a final sparQ or SPRI volume of 2X sparQ or SPRI.
- Transfer entire volume into a low-bind 1.5mL tube.
- Incubate 10 minutes at room temperature.
- Place tube on magnet and wait 2 minutes until solution is clear.
- Carefully remove and discard the supernatant.
- Add 400 µl 80% Ethanol to the tube without disturbing the pellet and stand for 30 seconds (only one Ethanol wash).
- Carefully remove and discard the ethanol wash.
- Centrifuge tube briefly and return it to magnet.
- Remove and discard any remaining ethanol.
- Resuspend beads in 50 µl water.
- Perform another round of 2X sparQ or SPRI purification by adding 100 µl sparQ or SPRI reagent directly onto resuspended beads.
- Mix by pipetting, and incubate 10 minutes at room temperature.
- Place tube on magnet and wait 2 minutes until solution is clear.
- Carefully remove and discard the supernatant.
- Add 200 µl 80% Ethanol to the tube without disturbing the pellet and stand for 30 seconds (1st Ethanol wash).
- Carefully remove and discard the ethanol wash.
- Add 200 µl 80% Ethanol to the tube without disturbing the pellet and stand for 30 seconds (2nd Ethanol wash).
- Carefully remove and discard the ethanol wash.
- Centrifuge tube briefly and return it to magnet.
- Remove and discard any remaining ethanol and allow the beads to air dry for 2 minutes (do not over dry beads).
- Resuspend beads in 90 µl water.
- Pipette mix vigorously and incubate at room temperature for 5 minutes.
- Place tube on magnet and transfer clear supernatant into two PCR tubes.
2) Amplify ADT sequencing library
Prepare 100µL PCR reaction with purified ADTs:
- 45 µl purified ADT fraction (also contains Hashtags, but they are not amplified at this step)
- 50 µl 2x KAPA HiFi PCR Master Mix
- 2.5 µl Truseq Small RNA RPIx primer (containing i7 index) 10 µM.
- 2.5 µl P5 oligo at 10 µM depending on application:
- For Drop-seq use P5-SMART-PCR hybrid oligo.
- For 10X use SI PCR oligo.
|
Cycling conditions:
|
| 95°C |
3 min |
|
|
| 95°C |
20 sec |
| |
|
| 60°C |
30 sec |
| |
6-10 cycles |
| 72°C |
20 sec |
| |
|
| 72°C |
5 min |
|
|
3) Amplify HTO sequencing library:
Prepare 100µL PCR reaction with purified Hashtags:
- 45 µl purified Hashtag fraction (also contains ADTs, but they are not amplified at this step)
- 50 µl 2x KAPA HiFi PCR Master Mix
- 2.5 µl Truseq DNA D70x_s primer (containing i7 index) 10 µM.
- 2.5 µl P5 oligo at 10 µM depending on application:
For Drop-seq use P5-SMART-PCR hybrid oligo.
For 10x use SI PCR oligo.
|
Cycling conditions:
|
| 95°C |
3 min |
|
|
| 95°C |
20 sec |
| |
|
| 64°C |
30 sec |
| |
8-12 cycles |
| 72°C |
20 sec |
| |
|
| 72°C |
5 min |
|
|
4) Purify PCR product using 1.6X sparQ or SPRI purification by adding 160 µl sparQ or SPRI reagent.
- Incubate 5 minutes at room temperature.
- Place tube on magnet and wait 1 minute until solution is clear.
- Carefully remove and discard the supernatant.
- Add 200 µl 80% Ethanol to the tube without disturbing the pellet and stand for 30 seconds (first Ethanol wash).
- Carefully remove and discard the ethanol wash.
- Add 200 µl 80% Ethanol to the tube without disturbing the pellet and stand for 30 seconds (second Ethanol wash).
- Carefully remove and discard the ethanol wash.
- Centrifuge tube briefly and return it to magnet.
- Remove and discard any remaining ethanol and allow the beads to air dry for 2 minutes.
- Resuspend beads in 20 µl water.
- Pipette mix vigorously and incubate at room temperature for 5 minutes.
- Place tube on magnet and transfer clear supernatant to PCR tube.
5) ADT and HTO libraries are now ready to be sequenced.
- Quantify libraries by standard methods (QuBit, BioAnalyzer, qPCR).
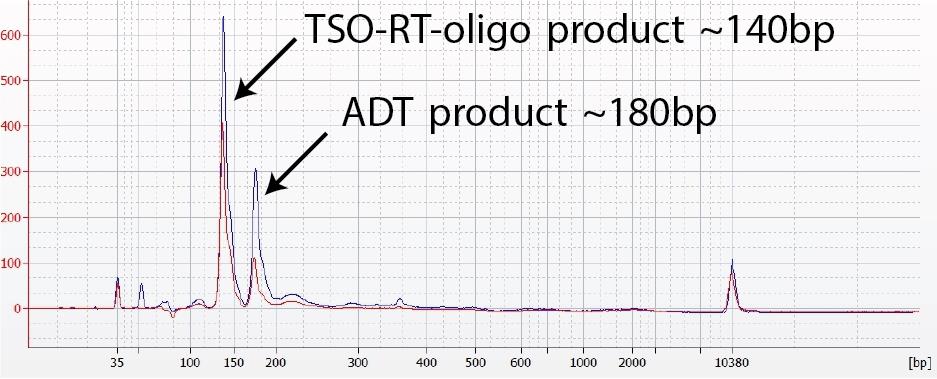
- ADT and Hashtag libraries will be around 180bp (Figure 1 and 2).
IV) Sequencing CITE-seq libraries:
We estimate that an average of 100 molecules per ADT or HTO per cell is sufficient to achieve useful information. The number of reads required to obtain 100 molecules depends on the complexity of the sequencing library (e.g. duplication rate). ADT, HTO and cDNA sequencing libraries can be pooled at desired proportions. To obtain sufficient read coverage for both libraries we typically sequence ADT at 10% and HTO libraries at 5% of a lane and cDNA library fraction at 85% of a lane (HiSeq2500 Rapid Run Mode Flowcell).
Please visit https://www.biolegend.com/totalseq for detailed information.
Oligos required for ADT and HTO library amplification:
• Drop-seq P5-SMART-PCR hybrid primer (for Drop-seq only) 5’AATGATACGGCGACCACCGAGATCTACACGCCTGTCCGCGGAAGCAGTGGTATCAACGCAGAGT*A*C
• 10x Genomics SI-PCR primer (for 10x Single Cell Version 2 only) 5’AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGC*T*C
• ADT cDNA PCR additive primer 5’CCTTGGCACCCGAGAATT*C*C
• HTO cDNA PCR additive primer 5’GTGACTGGAGTTCAGACGTGTGC*T*C
• Illumina Small RNA RPI1 primer (for ADT amplification; i7 index 1, Oligonucleotide sequences © 2015 Illumina, Inc) 5’CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCCTTGGCACCCGAGAATTC*C*A
• Illumina TruSeq D701_s primer (for HTO amplification; i7 index 1, shorter than the original Illumina sequence) 5’CAAGCAGAAGACGGCATACGAGATCGAGTAATGTGACTGGAGTTCAGACGTGT*G*C
* indicates a phosphorothioate bond
B indicates C or G or T; not A nucleotide

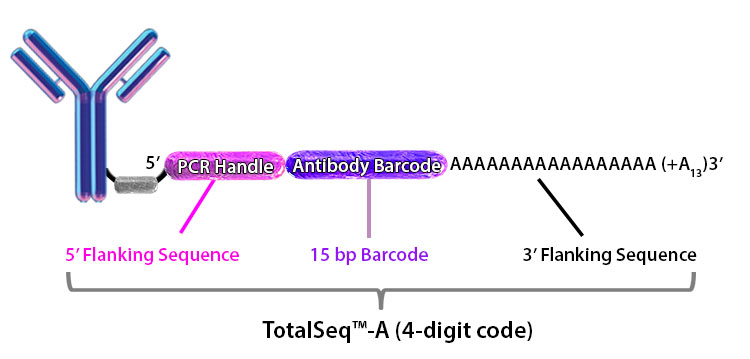
Figure 1. ADT or HTO library structure.
A.
 |
B.
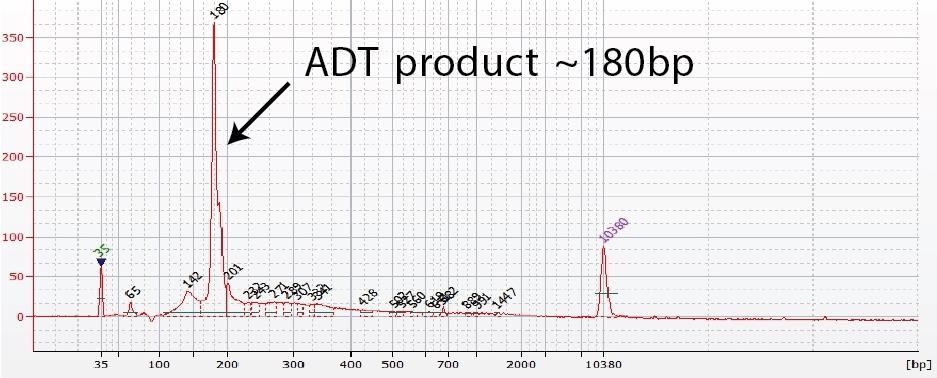 |
Figure 2. ADT (or hashtag) library verification. (a) A TSO-RT-oligo product (~140 bp) can be amplified during the ADT PCR by carryover primers from cDNA amplification. The product will not cluster but will interfere with quantification. Sequential 2X sparQ or SPRI purification of the ADT fraction after cDNA amplification reduces carryover of primers from cDNA amplification, and minimizes the amplification of this product during ADT-library amplification. To further enrich for ADT (or HTO) specific product the purified ADT library can be reamplified for ~3 additional cycles. (b) A clean ADT (or HTO) library will contain a predominant single peak at around 180 bp.
 |
Figure 3. Verification of ADT and hashtag libraries. ADT and Hashtag libraries are very similar in size ~180bp but should appear as distinguishable products on a High Sensitivity Bioanalyzer, where the Hashtag library appears a few nucleotides larger compared to the ADT library. |